Learn the Language of Proteins Efficiently with ELECTRA
The Natural Language Processing algorithm BERT has shown remarkable usefulness for biological sequence processing tasks. RoseTTAFold, a revolutionary software tool for accurate protein structure prediction, uses the BERT algorithm during training. Training BERT is expensive and time-consuming, requiring access to a GPU computing cluster. ELECTRA is an algorithm like BERT but is orders of magnitude faster and more computationally efficient. With a single PC and gaming GPU, we train ELECTRA to “learn the language of protein sequences” and process protein sequence data. ELECTRA achieves strong results on multiple benchmarks despite using far less compute and far fewer training steps compared to competing deep-learning methods. Corroborating the natural Language Processing literature, we show that using ELECTRA instead of BERT for biological sequence processing tasks saves time and resources, or learns better given the same compute budget. The results suggest protein structure prediction algorithms like RoseTTAFold should use the ELECTRA algorithm for training rather than BERT. The code and pretrained model weights are available at https://github.com/Joseph-Vineland/ProteinELECTRA
BERT and Natural Language Processing
The aim of Natural Language Processing (NLP) is to give computers the ability to understand text the same way humans can. The BERT algorithm has shown record breaking, state-of the art performance across a wide variety of NLP benchmark tasks. It can grasp syntax and semantics, and can interpret intent and sentiment. Jay Alammar has great visual guides to help you understand how BERT works.
https://jalammar.github.io/illustrated-transformer/
https://jalammar.github.io/illustrated-bert/
Details of the algorithm can be understood by referring to the original paper.
https://arxiv.org/pdf/1810.04805.pdf
In summary, there are two steps to deploying BERT, pre-training and fine-tuning.
- Pre-training: Using a large corpus of unlabeled text, such as all of Wikipedia or 50 000 books, 15% of the words are randomly masked and the model is trained to predict the masked words based on the unmasked words nearby. For instance, “The city of New York is in the [ ] States of America.” From experience, a human reader knows that the missing word is “United.” Likewise, the BERT model learns to predict the missing word from experience. In this way, the BERT model learns to grasp patterns and rules within the language, and it also learns a great amount of general knowledge about the world. The process is repeated iteratively, with different words being masked each time. This is called “self-supervised” learning, since the data is unlabeled text.
- Fine-tuning: The pre-trained BERT model from step 1 is further trained with labeled data to perform a specific task. For instance, using a set of emails labeled as “spam” or “not spam”, the pre-trained model is trained further or “fine-tuned” to be able to read emails and classify them as “spam” or “not spam.” This is called “supervised” learning, since the data is labeled text examples. Although in theory the pre-training step is unnecessary for classification/regression tasks, it has been shown that pre-training makes a large positive impact in the predictive performance of the model. In fact, world records have been set for machine learning benchmarks using only a few dozen labeled examples, due to the power of pre-training models. The knowledge gained from pre-training on a massive unlabelled dataset is successfully “transferred” when learning to perform different classification and regression tasks. Therein lies the power of BERT.
BERT for Biology
We can apply BERT to protein sequence data and DNA sequence data, just like we can with human language data. Just like with human language, we want to give computers the ability to “understand” the language of protein sequences. From the sequence of letters alone, the computer should understand the message and the intent AKA the protein’s attributes, structure, function, and overall purpose in the wider context of the living organism.
“Like human language, protein sequences can be naturally represented as strings of letters. The protein alphabet consists of 20 common amino acids (AAs) (excluding unconventional and rare amino acids). Furthermore, like natural language, naturally evolved proteins are typically composed of reused modular elements exhibiting slight variations that can be rearranged and assembled in a hierarchical fashion. By this analogy, common protein motifs and domains, which are the basic functional building blocks of proteins, are akin to words, phrases and sentences in human language. Proteins are a natural fit for NLP methods. Another central feature shared by proteins and human language is information completeness. From an information-theory perspective, the protein’s information (e.g. its structure) is contained within its sequence.” - Ofer et al. 2021
Protein language syntax is learnable like human language syntax due to evolution. “The space of naturally occurring proteins occupies a learnable manifold. This manifold emerges from evolutionary pressures that heavily encourage the reuse of components at many scales: from short motifs of secondary structure, to entire globular domains.” – M. AlQuiraishi
Predicting protein structure from amino acid sequence is an important step in understanding and defining gene function. Recently programs such as RoseTTAFold have shown remarkable success in predicting protein structures based on the amino acid sequence. The accuracy of these deep learning methods has transformed protein structure biology and revolutionized small molecule drug discovery, protein design, and protein interaction modeling. A BERT approach was used when training RoseTTAFold. “In the original training of RoseTTAFold for structure prediction a small fraction (15%) of tokens in the multiple sequence alignment are masked, and the network learns to recover this missing sequence information in addition to predicting structure.” -Wang et al. 2021. The BERT algorithm has also been used successfully for many other biological sequence processing tasks. It regularly outperforms all previous approaches.
The downside with BERT is that the pre-training step is computationally expensive in the extreme, requiring access to a GPU computing cluster for extended periods of time. This restricts access and prevents many from teaching BERT to learn and understand the language of protein sequences from unlabeled data. Although it is possible to download a pre-trained model from someone else, the end-user then has no choice in the model parameters or the pre-training data used. Furthermore, machine learning is an iterative process, so trial-and-error testing with different learning parameters becomes impossible when the training process is so computationally expensive. The amount of unannotated protein data is growing at an exponential rate. There is an increasing need for efficient computational methods to extract information from unlabeled sequences.
ELECTRA is a better BERT
ELECTRA is like BERT but much faster and computationally efficient. Here is a brief explanation of how ELECTRA works.
- Pre-training: For ELECTRA pre-training, ~15% of the words are replaced rather than masked, and the model is trained to identify which words have been replaced. The replaced words are plausible but incorrect alternatives, as part of the ELECTRA model is simultaneously learning to generate reasonable sounding replacements.
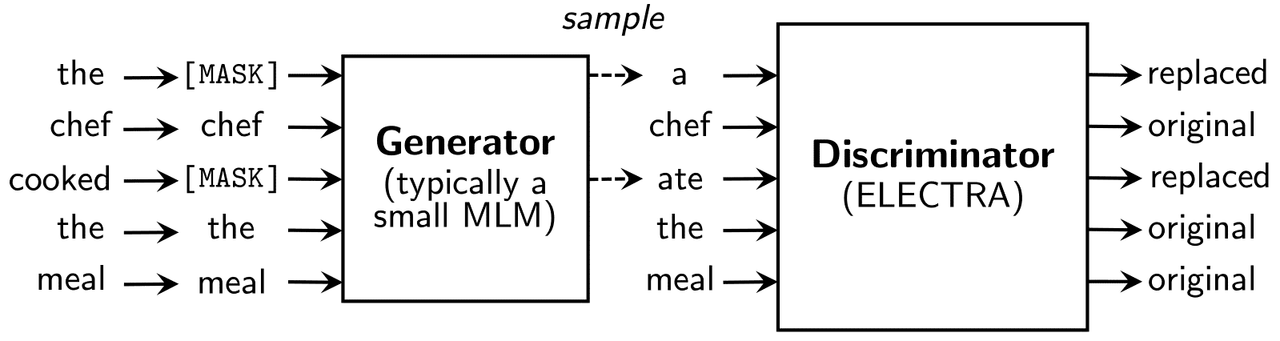
- Fine-tuning: The fine-tuning step for ELECTRA is the same as BERT.
The ELECTRA approach is more efficient partly because the task of learning to predict which words have been replaced is applied to all the words in the training data, not just 15% of them like the masked word BERT model. “ELECTRA needs to see fewer examples to achieve the same performance because it receives more training signal per example. At the same time, the ELECTRA approach results in powerful representation learning of language, because the model must learn an accurate representation of the data distribution in order to solve the task.” (From https://ai.googleblog.com/2020/03/more-efficient-nlp-model-pre-training.html)
Although similar to BERT, it has yet to be tested how well ELECTRA can be taught to correctly interpret the meaning of protein sequences like it can with human language word sequences. With a single GPU and a desktop computer, we show that ELECTRA can be used to successfully learn the language of proteins (pre-training) and apply the knowledge gained to different protein sequence processing tasks (fine tuning).
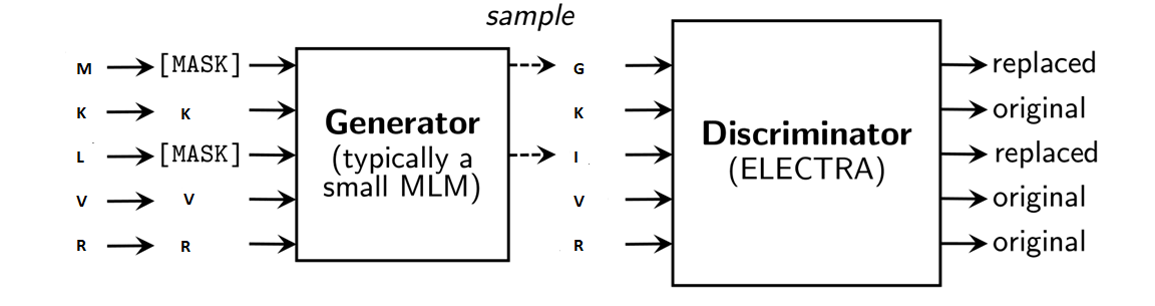
As shown above, we will apply ELECTRA to sequences of amino acids to learn the language of proteins.
Testing ELECTRA
The Tasks Assessing Protein Embeddings (TAPE) are standardized benchmark protein datasets used to evaluate the performance of protein sequence processing algorithms. There are five TAPE tasks spread across different domains of protein biology - secondary structure prediction, contact prediction, remote homology detection, fluorescence landscape predictions, and stability landscape prediction. For a full description of the different TAPE tasks, see the original publication. https://arxiv.org/pdf/1906.08230.pdf . The pre-training corpus consists of ~32.5 million protein sequences. For fine-tuning, each benchmark TAPE task has a training, validation, and test set. The TAPE data is curated and standardized to ensure effective methods for learning the TAPE tasks will translate to success in real-life scenarios. Researchers across the world use the same TAPE datasets for pre-training, fine-tuning, and testing, so they can directly compare the performance of different protein modeling algorithms.
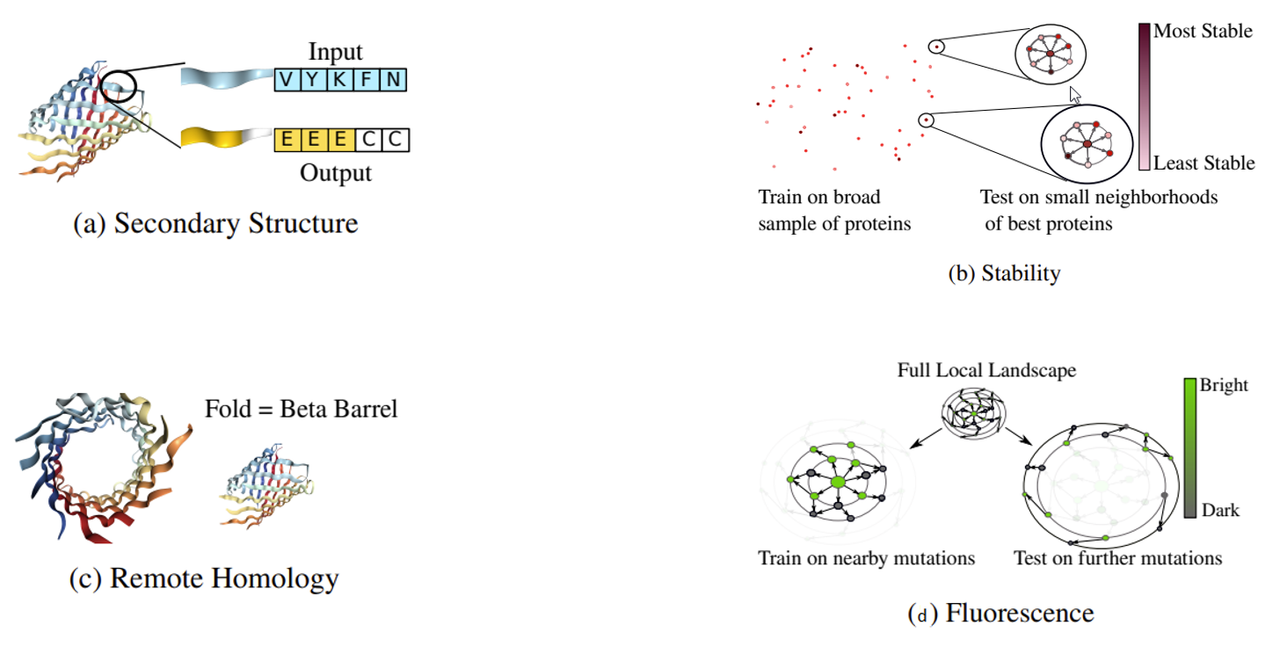 Images showing some of the TAPE tasks.
Images showing some of the TAPE tasks.
Here are the number of sequences in the TAPE data sets.
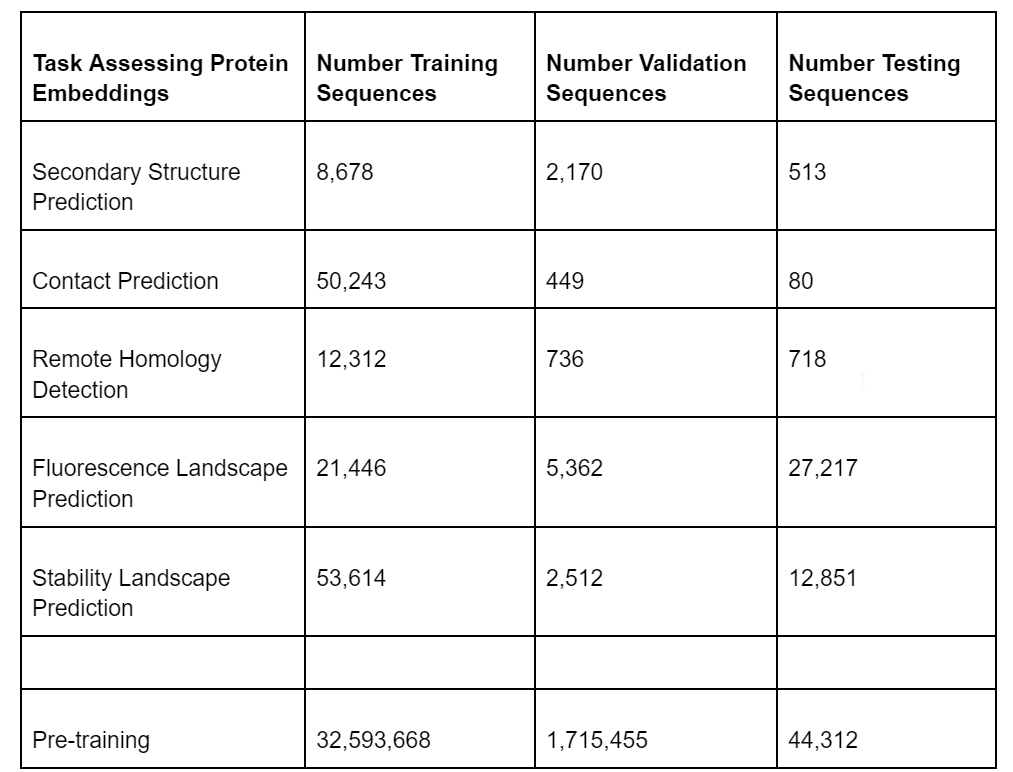 Table 1: Number of protein sequences in the TAPE benchmark datasets
Table 1: Number of protein sequences in the TAPE benchmark datasets
We use the TAPE benchmarks to evaluate the performance of ELECTRA. For protein sequences, we treat each individual amino acid as a word or “token.” There are 20 possible “words” in the language when we define the vocabulary this way. We pre-train ELECTRA with the TAPE pre-training “train” corpus for 3 epochs (passes over the dataset). We used the ELECTRA-Base sized model. The default ELECTRA-Base model parameters were used except for learning rate (5e-6) and batch size (4).We then take the pre-trained model and fine-tune it for four of TAPE tasks (independently) utilizing the “train” datasets. We omit the “Contact Prediction” task. The same fine-tuning parameters were used for each TAPE task. The default ELECTRA-Base model fine-tuning parameters were used except for learning rate (5e-6) , batch size (4), and number of epochs (5). The fine-tuning parameters were chosen via iterative testing to see what would give the best predictive performance on the “validation” sets. Finally, the fine-tuned models were tested using the “test” sets for each investigated TAPE task. Here are the results for ELECTRA on the TAPE benchmark tasks.
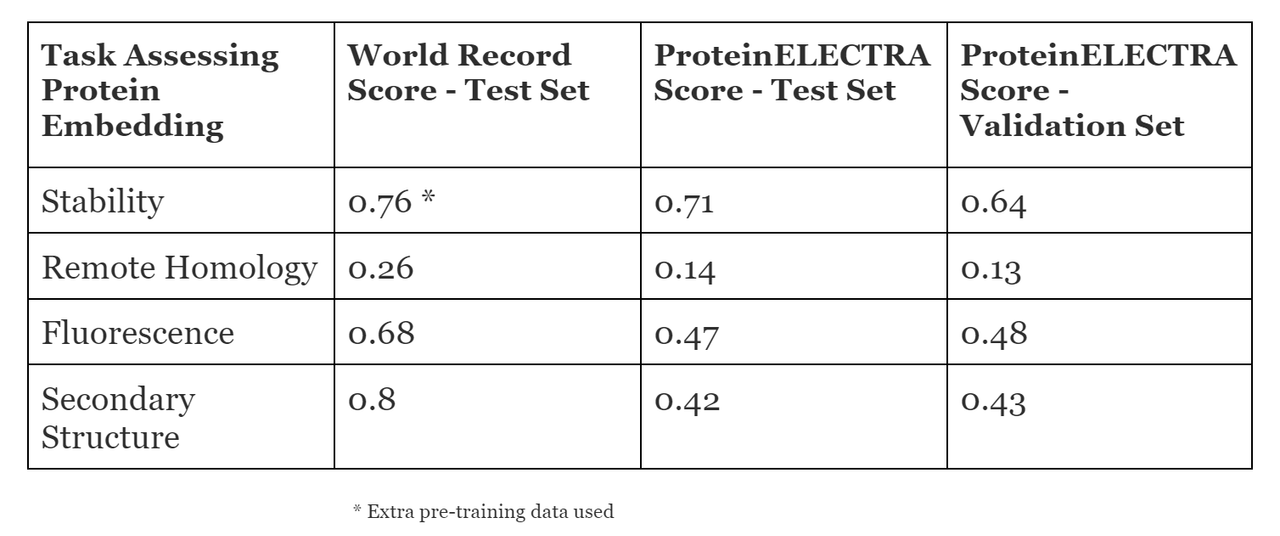
Pre-training took ~2.5 months with NVIDIA GeForce RTX 2080 Ti. After pre-training on the TAPE “pre-training” dataset, we evaluated ELECTRA’s performance using four TAPE benchmark tasks. (We omit the contact prediction task). Fine-tuning ELECTRA to perform a TAPE task takes ~20 minutes up to ~ 3 hours, depending on the task.We compare the performance of ELECTRA to state-of-the-art sequence models which had been evaluated on the same benchmarks. These are the highest scores for the benchmarks we could find in the literature, so we refer to them as the “World Record” scores. We observed that ELECTRA performs competitively with state-of-the-art sequence models despite using far less compute and training steps, particularly in the “Stability” benchmark. We note that the same fine-tuning parameters work well for all evaluated TAPE tasks, showing that ELECTRA makes an effective “out-of-the-box” model.
Discussion
ELECTRA can learn to understand the biological meaning of protein sequences efficiently and effectively. We show that ELECTRA can be used to extract information from unlabeled protein databases in a way that doesn’t require access to a special GPU computing cluster. Someone with a single gaming GPU can teach ELECTRA to learn the language patterns of protein sequences and apply that knowledge to elucidate biological processes. We show that using an ELECTRA approach instead of BERT to train protein structure prediction algorithms like RoseTTAFold would result in saving lots of time and resources. Or, it would achieve substantially more learning for the same cost. With the power of ELECTRA’s “transfer learning” (pre-training -> fine-tuning), one has the capability to predict the protein’s attributes, structure, function, and overall purpose in the wider context of the living organism from the amino acid sequence alone.
While we haven’t broken the world records of specialized algorithms, we show that ELECTRA is a very fast learner and requires minimal pre-training to pick up “the language of proteins.” It achieves competitive results with far less compute and training steps, especially when compared against other transformer models. For some tasks, traditional non-deep-learning methods are still superior to BERT or ELECTRA. However, ELECTRA is more flexible than these traditional methods and can be applied successfully to a wide variety of different tasks so its performance is overall better. Deploying ELECTRA for learning the language of proteins is a huge increase in computational efficiency compared to what’s been done previously. We’re doing it with one graphics card. Using ELECTRA’s sequence tagging routines for protein secondary structure prediction is a novel way of applying NLP techniques to biological sequence analysis. Pre-training an ELECTRA-Base sized model on one graphics card is possible, with some tweaks. (Use batch size of 4 and a low learning rate). This insight is useful for machine learning in general. In the future, perhaps we should explore incorporating Gene Ontology into the pre-training process and use a custom network architecture like ProteinBERT, to achieve better learning for protein sequence tasks.
Conclusion
In conclusion, the results suggest biological sequence processing protein structure prediction algorithms like RoseTTAFold should use the ELECTRA algorithm for training rather than BERT.
If you would like to try out ELECTRA for protein sequences, see the GitHub: https://github.com/Joseph-Vineland/ProteinELECTRA
Thank you to Jenny Crick for the writing instructions, advice, and encouragement.